

Researchers Sequence Dark Matter of Life
San Diego, Sept. 19, 2011 -- Researchers led by Pavel Pevzner, director of Calit2's Center for Algorithmic and Systems Biology (CASB), and Roger Lasker of the J. Craig Venter Institute, have developed a new method to sequence and analyze the dark matter of life -- the genomes of thousands of bacteria species previously beyond scientists' reach, from microorganisms that produce antibiotics and biofuels to microbes living in the human body.
|

"This part of life was completely inaccessible at the genomic level," said Pavel Pevzner, a computer science professor at the Jacobs School of Engineering at UC San Diego and a pioneer of algorithms for modern DNA sequencing technology.
Pevzner, in collaboration with UC San Diego mathematics professor Glenn Tesler and computer science postdoctoral researcher Hamidreza Chitsaz, developed an algorithm that dramatically improves the performance of software used to sequence DNA produced from a single bacterial cell. These programs traditionally recover 70 percent of genes.
"The new assembly algorithm captures 90 percent of genes from a single cell. Admittedly, it is not 100 percent. But it's almost as good as it gets for modern sequencing technologies: today biologists typically capture 95 percent of genes but they need to grow a billion cells to accomplish it," said Tesler.
Bacteria play a vital role in human health. They make up about 10 percent of the weight of the human body and can be found anywhere from the stomach to the mouth. Some, like E. coli, can wreak havoc. Others help us digest. Yet others, recent studies have found, can change the way we behave by, for example, tricking us into eating more than we need. That's why it is crucial to analyze bacteria's genomes, which in turn help scientists understand bacteria's behavior.
|
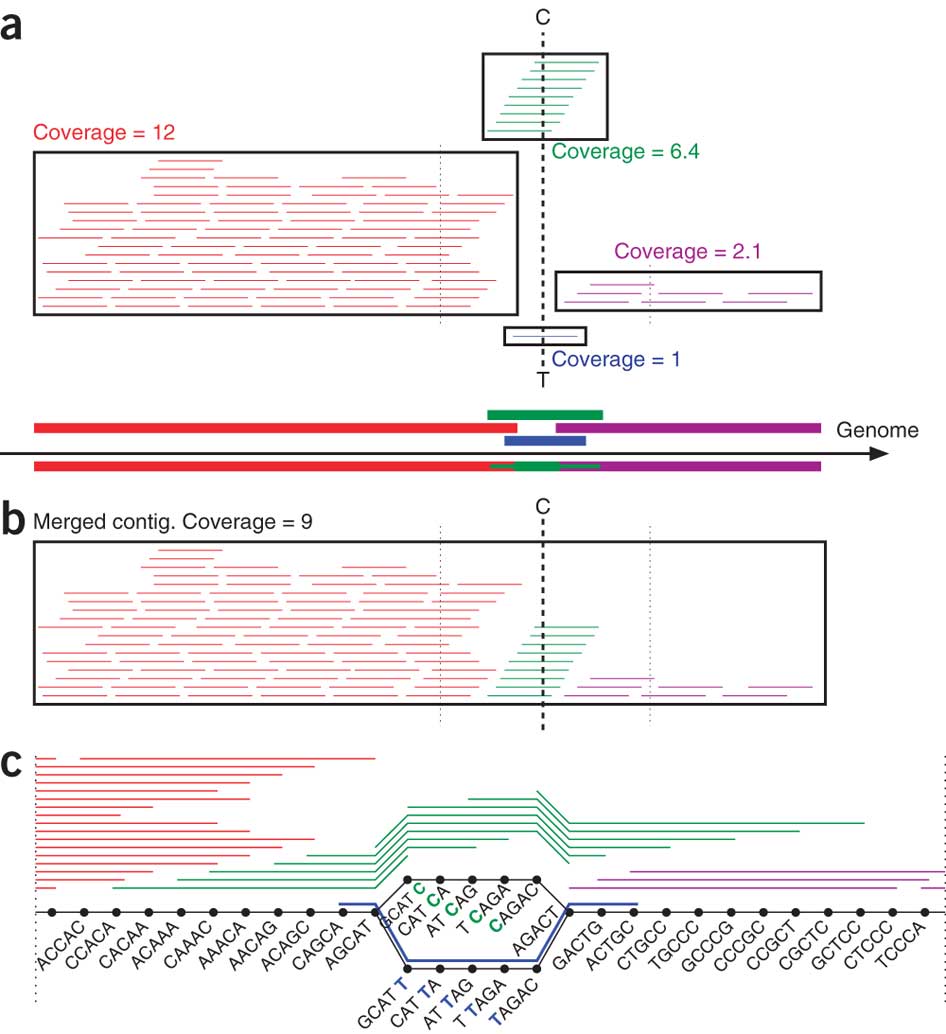
Enter Multiple Displacement Amplification (MDA) technology, developed about a decade ago by Professor Roger Lasken, now at the Venter Institute and co-author of the Nature Biotechnology study. MDA can be used on bacteria that can't be cultured in the lab. The technology is the equivalent of a copy machine that starts from a single cell and makes copies of fragments of its genome until it produces the equivalent of one billion cells. In 2005, Lasken and colleagues used MDA to sequence DNA produced from a single cell for the first time with funding from the Department of Energy.
However, while MDA is an ingenious cellular copy machine, it gives sequencing software programs a hard time. The DNA copies that MDA makes carry various errors and are not amplified uniformly: some pieces of the genome are copied thousands of times, and others only once or twice. Modern sequencing algorithms aren't equipped to deal with these disparities. In fact, they tend to discard bits of the genome that were replicated only a few times as sequencing errors, even though they could be key to sequencing the whole genome. The algorithm developed by Pevzner's team changes that. It retains these genome pieces and uses them to improve sequencing.
Researchers sequenced a single cell of E. coli with this method to verify the accuracy of the algorithm and recovered 91 percent of its genes, doing nearly as well as conventional sequencing from cultured cells. This provides enough data to answer many important biological questions, such as what antibiotics a species of bacteria produces. It also, for the first time, enables researchers to perform in-depth studies to figure out which proteins and peptides the bacteria living in human beings use to communicate with each other and with their host.
|

Pevzner's team is at work on a second-generation version of the algorithm. Lasken and his team plan to continue their work on improving MDA as well.
Lasken keeps a few hundred tubes filled with unsequenced bacteria in his laboratory at the Venter Institute in La Jolla, Calif. Each represents a bacterial terra incognita that scientists soon will explore using the method developed through the combined efforts of researchers at the UC San Diego Jacobs School of Engineering, the Venter Institute and Illumina.
"It's a very big step forward," Lasken said.
The research was partially supported by grants from the National Human Genome Research Institute and the Alfred P. Sloan Foundation and by a grant from the National Institutes of Health.
Efficient de novo assembly of single-cell bacterial genomes from short-read data sets, Nature Biotechnology (2011), by Hamidreza Chitsaz, Joyclyn L Yee-Greenbaum, Glenn Tesler, Mary-Jane Lombardo, Christopher L Dupont, Jonathan H Badger, Mark Novotny, Douglas B Rusch, Louise J Fraser, Niall A Gormley, Ole Schulz-Trieglaff, Geoffrey P Smith, Dirk J Evers, Pavel A Pevzner & Roger S Lasken. Published online September 18, 2011.
Related Links
Center for Algorithmic and Systems Biology
Nature Biotechnology
Jacobs School of Engineering
Computer Science and Engineering Department