

Evolution, Software and Microinversions: Building Better Phylogenetic Trees
San Diego, CA, December 19, 2006 -- Biologists will be able to reconstruct the process of evolution, determine relationships between species and build phylogenetic trees with greater accuracy thanks to a new method for identifying “microinversions,” which are extremely short strings of inverted nucleotides. This new work from researchers at UC San Diego and Brown University appearing this week in the early online edition of the Proceedings of the National Academy of Sciences (PNAS).
|

Microinversions - usually tens to thousands of base pairs in length - can only be detected if you have the exact nucleotide sequence of the same genomic region for all the species you are considering. Many recent studies have pointed to microinversions as large sources of genetic diversity that have not previously been characterized, and the new research from UCSD provides a more careful and accurate approach to identifying microinversions.
"As more fine-grained genomic data becomes available, microinversions will be increasingly important in understanding genetic diversity both between and within species," said Mark Chaisson, the first author on the paper and a Bioinformatics Ph.D. student from UCSD's Jacobs School of Engineering.
"This method might be able to provide evidence for the entire mammalian phylogeny, such as the presence of an Afrotheria clade," he said. A clade is a cohort of mammals that are all descended from a common ancestor, and DNA analysis indicates that Afrotheria includes nearly a third of all mammalian orders currently found in Africa and Madagascar -- including many species that are at risk of extinction.
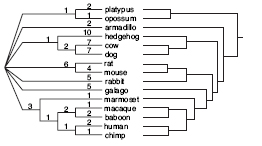
Using data from their microinversion detection technique - an open-source software system called InvChecker - the researchers reconstructed the phylogenetic tree for 15 mammals. This work largely confirmed the existing phylogenetic tree that connects these mammals.
|

"We've only looked for microinversions in 0.1 percent of the genomic sequence from several mammals, and we can already confirm many of today's ideas about the history of evolution. When similar analyses extend to one percent of the genomes under investigation, we'll have a 10-fold increase in data. This should shed light on splits between species that have been debated in molecular evolution," explained Pevzner.
"This microinversion detection method could be used for detecting human structural variants once we have the necessary data," explained Ben Raphael, a former postdoc in Pevzner's lab and now a professor of computer science at Brown University. Raphael is the second author on this paper and a former postdoctoral researcher at UCSD. Raphael helped establish CASB and is organizing the RECOMB Satellite Workshop on Computational Cancer Biology at Calit2 next September 16-18, 2007.
|
With InvChecker, the researchers analyzed the CFTR region in a collection of mammal species. CFTR is a heavily studied and highly conserved, gene-rich area of human chromosome 7 that is home to the cystic fibrosis gene.
"It's quite a subtle problem to find microinversions. Our goal is to use these tiny inversions to develop a history of species," said Pevzner.
The researchers also used InvChecker to study the specific differences between humans and chimpanzees. They found that 80 percent of the microinversions between humans and chimps that were proposed last year are, in fact, repeat-induced artifacts and not microinversions. The researchers also uncovered 167 human-chimp microinversions recently missed by scientists using software other than InvChecker.
"This finding doesn't change the conclusions between humans and chimps, but it does say that the detection of microinversion needs to be done carefully," said Chaisson. "InvChecker does a more careful job of comparing sequences than previous attempts to find microinversions."
|

"We are looking for orthologous sequences in reverse order that are surrounded by elements in forward order. That's a microinversion," said Chaisson.
Microinversions have certain advantages over other evolutionary signals used for studying evolution such as amino acid changes, Chaisson explained. "With microinversions, it's easy to develop evolutionary relationships between species and difficult to debate whether one species is inverted relative to another species."
With InvChecker and microinversions, researchers are not limited to comparing species that are evolutionarily close, as is the case when using other genomic features like repetitive sequences and deletions for phylogenetic analysis. The new process can also detect microinversions that are the result of convergent evolution and thus do not play a role in tracking evolution and defining phylogenies.
Once the researchers have the microinversion data, they use it to reconstruct phylogenies using an algorithm that attempts to move "back in time" by iteratively undoing microinversions and bringing the existing species closer to the ancestral mammalian genome.
Related Links
Proceedings of the National Academy of Sciences
Center for Algorithmic and Systems Biology
Media Contacts
Media Contact: Daniel Kane, 858-534-3262, dbkane@ucsd.edu