

UC San Diego and Genentech Scientists Develop Potentially Disruptive Antibody Sequencing Technology
San Diego, Dec. 18, 2008 -- Bioinformatics researchers at the University of California, San Diego and Genentech have developed a new, quicker way to sequence monoclonal antibodies – a process that is many times faster than the sequencing technology typically used by academic and industry researchers today.
|

While DNA sequencing technologies witnessed dramatic progress in recent years, protein sequencing has hardly changed in 50 years. As a result, today nearly all proteins are discovered using DNA rather than protein sequencing technology. While it works for most proteins (that are coded by DNA), it does not work for some important proteins, such as antibodies, that are not directly inscribed in genomes.
“Our new approach has the potential to be a disruptive technology for all protein sequencing applications,” said Nuno Bandeira, lead author on the paper and director of the new Center for Computational Mass Spectrometry (CCMS) at UC San Diego. “This project is a collaboration with Genentech, the leader in development of antibody-based drugs, and it illustrates the potential impact that this center and this technology can have on the biotech industry in California and around the world.”
|
Shotgun protein sequencing will be particularly useful when complementary DNA (cDNA) or the original cell line is not available, or if there is a need to verify the integrity and effectiveness of an antibody after the cell has undergone changes subsequent to the original sequencing.
“Antibodies are indispensable in biomedical research and they are widely used as diagnostic and therapeutic agents,” said Genentech’s Jennie Lill. “DNA sequencing is routinely used in the initial characterization of monoclonal antibodies, but subsequent mutations and other changes mean that further protein level analysis is needed. So it is critical to sequence the antibodies for a variety of reasons, from monitoring the integrity of the molecule, to troubleshooting performance in pre-clinical assays.”
Until now, the only viable option for sequencing an antibody has been a process known as Edman degradation, named for Swedish chemist Pehr Edman. (The technique was used in the sequencing of insulin, for which the Nobel Prize in Chemistry was awarded in 1958.) While Edman degradation remains a low-throughput and time-consuming approach, no fast substitute for this technique was found in the last half-century.
Bandeira and his colleagues proposed to substitute this ancient technique with protein sequencing based on mass spectrometry.
While mass spectrometry routinely is used to sequence short fragments of proteins (called peptides), no techniques for sequencing entire proteins were available until recently. The key bottleneck has been computing rather than experiment, since the challenge of protein assembly is a puzzle rivaling the complexity of DNA sequencing.
|
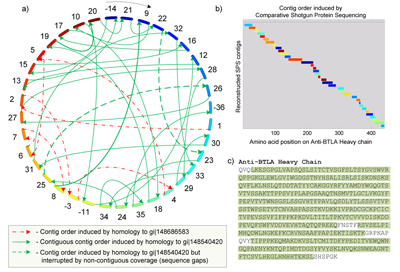
Bandeira offers a simple analogy for the algorithmic foundations of CSPS. “Imagine that the revised edition of a popular book has just been printed and that a competitor wishing to delay its release sneaks into the warehouse to shred all the books to pieces and destroys the original template,” observed Bandeira. “In this context, the SPS step allows one to reconstruct whole portions of the text by assembling snippets into sections and chapters (similar to a puzzle-solving approach), and the comparative step uses very old editions of the book to reorganize the parts back into a complete copy of the latest edition. By comparison with CSPS, competing protein-sequencing techniques are much more labor-intensive and would more closely resemble the process of asking the author to recite the whole book from memory.”
Replacing Edman degradation with CSPS enables sequencing at a fraction of the time. In addition, CSPS automatically detects post-translational modifications that might never have been observed with Edman degradation or even other mainstream peptide-identification strategies.
|

Concluded co-author Pavel Pevzner: “CSPS opens up many possibilities for sequence discovery in the biotech industry compared with traditional methods.”
In their paper, the bioinformatics researchers admit that while CSPS can readily handle small protein mixtures, more work is needed in order for the technique to fulfill its full potential for complete-proteome analyses. Ongoing research in UC San Diego’s Center for Computational Mass Spectrometry will focus on ways to improve the method’s efficiency, reliability and robustness.
Publication: ‘Automated de novo protein sequencing of monoclonal antibodies,’ by Nuno Bandeira , Victoria Pham, Pavel Pevzner, David Arnott and Jennie R. Lill. Nature Biotechnology, December 2008, v26, n12, pp1336-1338. The project was supported by National Institutes of Health grant NIGMS 1-R01-RR16522.
Related Links
Nature Biotechnology article: Automated de novo protein sequencing of monoclonal antibodies
Media Contacts
Doug Ramsey, 858-822-5825, dramsey@ucsd.edu